Biology REU 2015 Awardees

Sexual Dimorphism in Skates of the Genus Fenestraja
Bryan Kao (Columbia University)
Mentors: John Sparks and Christopher Martinez, Department of Ichthyology, Division of Vertebrate Zoology
The skates (Rajidae) are a diverse family of cartilaginous fishes, with many species that are currently understudied. However, recent work has shown that sexual dimorphism is a prevalent feature across the family. Many of these dimorphic characters appear in mature males and are not found in females. Changes in maturing males include increasing concavity of the anterior portion of their pectoral fins that causes the body to acquire a bell-like shape, appearance of alar thorns on their pectoral fins and malar thorns on their cheeks, and elongation of claspers (male reproductive organs). To learn more about the extent of sexual dimorphism in skates, a case study was performed on Fenestraja plutonia, a bathydemersal inhabitant of the western Atlantic that is typically found at depths of 290-750 meters. Fenestraja plutonia was an ideal subject for the study due to several reasons: 1) they exhibit strong sexual dimorphism in a number of traits, 2) specimens were readily available from the collections of the American Museum of Natural History and the Harvard Museum of Comparative Zoology, and 3) they are small and easy to work with. Forty-three specimens, representing males and females, adults and juveniles, were X-rayed and photographed. A combination of geometric morphometrics and traditional linear measurements were used to study dimorphism of the internal and external anatomy of F. plutonia. Additionally, a limited number of four other Fenestraja species were evaluated for comparison. Preliminary results point to significant differences in several morphological features between males and females, including the pectoral girdle, pelvic girdle, and jaws. This work provides useful information for future work on sexual dimorphism in skates and in other elasmobranchs.

3D Imaging of Hyoid Articulation in Elasmobranchs with Relation to Inner Ear Landmarks
Rachel Hutchins (University of Montana)
Mentors: John Maisey and Allison Bronson, Division of Paleontology
Sharks, rays, and skates collectively make up the elasmobranchs, a subgroup of the cartilaginous fishes or class Chondrichthyes. In many elasmobranchs, the hyoid arch (which helps support the jaws) is often conserved in its ancestral position, attaching to the cranium anteriorly to the glossopharyngeal (IX) nerve foramen. However, in the superorder Galeomorphii,the hyoid attachment is said to ‘extend anteriorly’. This has often been cited as a unifying trait for this seemingly disparate group that ranges from the pelagic Shortfin Mako Shark (Isurus oxyrinchus) to the bottom-dwelling Pygmy Ribbontail Catshark (Eridacnis radcliffei). My project was to test this supposition by comparing the extent of the articulation in a wide range of galeomorphs and other elasmobranchs. Lacking external cranial landmarks near the hyoid, inner ear morphology was used to qualitatively describe the point of articulation. CT scans of 20 elasmobranch species were manually segmented using digital reconstruction software. The relationships between hyomandibulae and inner ear structures were then visually analyzed and compared. While anterior extension was observed in most galeomorph species, sharks of the order Lamniformes exhibited poor hyoid calcification with the articulation far from the inner ear region. It has been previously suggested that in the order Orectolobiformes the hyoid may have migrated forward significantly, extending from the glossopharyngeal foramen across much of the otic capsule, and even entering the back of the orbit. We confirmed this, noting that while the ancestral articulation appears near the back of the horizontal canal, the anterior margin in Orectolobiformes is found closer to the utriculus. More surprisingly, the hyoid of Eridacnis radcliffei, a member of order Carcharhiniformes, is located completely anteriorly, beneath the utriculus and distant from the glossopharyngeal foramen. More research is required to determine whether this second condition is particular to the genus Eridacnis or exists in related taxa as well. There may be a connection between buccal pump respiration and the derived articulations, but this also remains to be examined.

Phylogenetic Position of the Extinct Metatherian Deltatheridium pretrituberculare and its Implications in Mammal Diversification
Devin K Hoffman (Appalachian State University)
Mentors: Michael J. Novacek and Paúl M Velazco, Division of Paleontology
Marsupials are a diverse group of mammals with approximately 350 extant species and many more extinct species. One of these extinct species, Deltatheridium pretrituberculare, known from the Late Cretaceous of Mongolia, is considered a member of the extinct Order Deltatheroida. Recent studies using small character datasets have recovered Deltatheridium as a basal metatherian. In order to test the affinities of Deltatheridium in a larger dataset, I added this taxon to the Mammal Tree of Life matrix built by O’Leary et al. (2013). The Mammal Tree of Life matrix is the largest phylogenetic study of mammals to date that includes both extant and extinct taxa. The matrix comprises 86 fossil and extant taxa, 4541 phenomic characters, and 27 nuclear genes. To add Deltatheridium to the matrix I generated digital images and CT reconstructions of several specimens, and then uploaded them to MorphoBank. Once all possible scorings were made, the updated matrix was run using PAUP* to obtain the most parsimonious trees. The resulting tree recovered Deltatheridium pretrituberculare as a basal metatherian in a well-supported monophyletic group. This result supports the original hypothesis of the placement of Deltatheroida as a basal metatherian. An unexpected result of the addition of Deltatheridium was the shift of leptictids, a group included within crown-group Placentalia in the original Mammal Tree of Life, to outside Placentalia. This underscores the argument in O’Leary et al. (2013), that the placental radiation was explosive, occurring after the K-Pg extinction event. Leptictids remain a monophyletic group and the node is well-supported. The Mammal Tree of Life project is still ongoing, with many more taxa needing to be added. More Mesozoic taxa need to be scored to determine the origination of many higher order groupings, including the timing of the Eutheria-Metatheria split. As more fossil specimens are uncovered, previous scorings can be revised and improved upon, giving a more accurate view of the diversification of mammals.
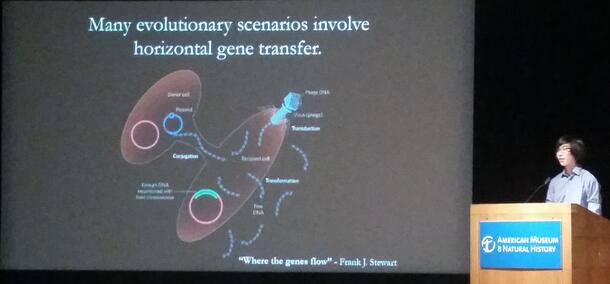
Construction and Diagnosis of Hybridization Networks as an Alternative to Trees in Phylogenetics
Michael Miyagi (The University of Texas at Austin)
Mentor: Ward Wheeler, Division of Invertebrate Zoology
In many microbial taxa wherein horizontal gene flow is a significant factor, graphs with a restriction of a single parent per node, commonly known as trees, are insufficient to describe evolutionary history. As a more general structure, networks can include reticulation events that allow any given node to have arbitrarily many ancestors and descendants, modeling varied gene ancestry in a way trees cannot. In order to objectively compare tree solutions with network solutions, we developed a software pipeline that takes raw gene sequences and generates a tree for each sample and the sum sequence using POY. Using an edge-removing heuristic, we compiled these directed output trees into a directed display network in polynomial time, a huge decrease in time complexity as compared to current algorithmic approaches to minimum hybridization networks. By assigning increased costs to reticulate edges, we compensated for the naturally decreased tree cost afforded by soft-wired networks and were able to differentiate between datasets which were better described by trees versus networks using the graph evaluation software PCG. Using local collinear blocks of the genomes of Vibrio varieties, segments from influenza, and simulated genomes, we tested the robustness and speed of this pipeline, and found that it was able to significantly reduce analysis time for large sets of input trees.

Comparative Scale Morphology and Ultrastructure Across Insect Lineages, with Special Reference to Weevils (Coleoptera: Curculionoidea)
Kyle DeMarr (Cornell University)
Mentors: Dave Grimaldi and Steve Davis, Division of Invertebrate Zoology
Structuring of the cuticle underlies morphological complexity and diversity throughout the insects. A common motif of cuticle formation recurs in the form of scales—structurally variable setal modifications secreted by a single epithelial cell. Scales are widespread among insects, having evolved multiply in Apterygota, Palaeoptera, Polyneoptera, Paraneoptera, and Holometabola. This study investigates the breadth of scale morphologies via scanning and transmission electron microscopic imaging to draw conclusions as to the patterns of convergence of these morphological features. Although studied extensively in lepidopterans, the lack of scale-less basal representatives in this order makes it difficult to address questions pertaining to scale origins. To better understand the inception of scale development and variation derived in the process of seta-scale transformation, their evolutionary progression was herein informed through multi-ordinal sampling. From this, the curculionoid lineage, containing weevils and their kin, rose as a model for scale evolution—elucidating a progression from the setae and simple, columnar scales of Nemonychidae to the elaborate, structurally diverse scales of the more derived Curculionidae. Similarly, preliminary TEM imaging of 23 taxa from representative subfamilies of Curculionoidea yielded a morphocline in the scale interior from loosely packed spherules to dense networks of cuticular bridges and photonic crystalline constructions. Developmental studies of the model fly Drosophila melanogaster have implicated several genes in setal patterning and ontogeny; to further consider scale development from both evolutionary and genetic perspectives, RNA interference was incorporated to determine the function of two such genes, Hairless and split ends, in scale formation of the rice weevil, Sitophilus oryzae. Insect scales serve as an example of extreme convergence and their study and description facilitates future efforts to comprehend mechanisms of pattern formation in natural systems, in addition to informing phylogeny and the field of biomimetic design.<hr style="border:0;width:96%;background-color:#D9D9BF;color:#D9D9BF;height:1px;">

Consideration of a Contagious Clam Cancer: Coevolution or eCological Co-incidence
Ashley Paynter (SUNY Binghamton)
Mentor: Mark Siddall, Division of Invertebrate Zoology
Previous work by Metzger et al. (2015) proved that a high copy number of the retrotransposon called Steamer, originally found in Mya arenaria (soft-shelled clam, or steamer clam), is the cause of a contagious and fatal leukemia-like neoplasm in bivalves. In order to better understand whether the retrotransposon is unique to soft-shelled clams or is phylogenetically versus ecologically distributed among related bivalves, we investigated the presence of this retrotransposon in other species of bivalves that had been stored in the Museum's Ambrose Monell Cryo Collection. Additional fresh samples of Mya arenaria from the Long Island Sound, and various bivalves from a local fish market called Citarella were obtained. Multiple methods were used for DNA isolation. A mollusk specific kit with a phenol:chloroform:isoamyl step yielded better PCR products relative to conventional DNeasy kits. Using Ready2Go PCR beads, presence of the retrotransposon was confirmed in locally collected soft-shelled clams, in a sample of Ensis directus (Atlantic Razor clam) in the AMCC from the North Sea, as well as from commercially available Ensis directus from Maine. Modification of the amplification protocol to include Taq Gold yielded additional evidence of the retrotransposon in Macoma balthica (the Baltic clam) in the AMCC from the North Sea. Steamer sequences were unique to each species of bivalve in which it was discovered, though nucleotide differences were few. In addition to the expected complete-length amplicon, several isolates from Ensis directus exhibited retrotransposon sequences missing a 109-nucleotide region flanked by an inverted repeat suggestive of secondary structural importance. That the retrotransposon was not found in taxa closely related to the marine Atlantic Mya arenaria or Ensis directus (i.e., brackish water Dreissena polymorpha and Pacific Ensis macha) suggests that horizontal transmission in overlapping ecological habitats may be a better indication of the prevalence of the retrotransposon than phylogeny. Higher sequence diversity in Ensis directus may be evidence that Atlantic razor clams were the ultimate source of infections. It is worrisome that Baltic clams are infected in the North Sea where Ensis directus is invasive.

Barcoding a Leech infestation at Lake Minnewaska State Park
Na’ta’ne Miles (Northwest Indian College, Department of Environmental Science)
Mentor: Mark Siddall, Division of Invertebrate Zoology
Lake Minnewaska State Park Reserve is located in Ulster County, New York in the scenic region of Shawangynk Mountain ridge. The popular beach at Lake Minnewaska was closed off to the public in June, 2014 due to an infestation of unknown species of leech with a propensity for attaching to swimmers. In 2012, eutrophication coincided with the event of an illegal introduction of golden shiners (bait fish) and large-mouth bass. In order to determine the identity and possible origin of the leeches in Lake Minnewaska, we searched for leeches from the underside of rocks and submerged branches on the shores of Lake Minnewaska, Lake Awosting and the nearby Lake Mohonk in July 2015. Leeches were tentatively identified by external anatomy and through DNA barcoding with mitochondrial cytochrome oxidase-1 (cox1). The leeches infesting Lake Minnewaska were morphologically consistent with Helobdella modesta. Leeches matching the description of Erpobdella punctata, Helobdella modesta and the sanguivorous Placobdella picta were found in Lake Mohonk. No leeches were found in Lake Awosting. Amplification and sequencing of cox1 corroborated only the identity of Erpobdella punctata while suggesting an as-yet undetermined species of Placobdella. Phylogenetic analyses revealed several unrelated groups broadly distributed in North America, Europe and Asia, all matching the description of H. modesta and H. stagnalis, but grouping without obvious relationship to geography. Those infesting Lake Minnewaska were most similar to specimens from Pennsylvania and Oregon, and that those from Lake Mohonk were sister to the foregoing. We infer that the leeches in Lake Minnewaska are naturally occurring to the region. With the occurrence of eutrophication of the lake, it is possible that the changes in the ecosystem have enabled Helobdella modesta to thrive due to a rise in benthic food sources – (i.e.; Oligochaeta/Oligochetes). We note that the attachment to humans while swimming in the lake is in fact due to a behavioral characteristic of the leech in question, and despite the inaccuracies from media coverage Helobdella species are not blood-feeding, and carry no known health threat to humans.

How a Jack of All Trades Can Be a Master of Many: the Transcriptomic Basis of Host Plant Use by a Generalist Pest, Chloridea virescens (Lepidoptera: Noctuidae)
Kelsey Payne (CUNY-LaGuardia Community College)
Mentors: Sara Oppenheim and Robert DeSalle, Division of Invertebrate Zoology
Understanding the molecular basis of ecological adaptation is something of a holy grail for modern evolutionary biologists. While progress has been made in determining the genetic basis of variation in ecologically relevant traits with Mendelian inheritance patterns, many traits of interest are complex and involve quantitative variation across a range of phenotypes. The evolution of host plant range amongst herbivorous insects is such a trait, depending on the ability of insects to find, accept, and grow upon appropriate host plants. I examined the performance of a generalist pest insect, Chloridea virescens, on a chemically diverse subset of its many host plant families to determine whether different genes are used to cope with different host plant defenses. Young caterpillars were fed on host plant material for 8-12 days and their behavior and growth were recorded. Midgut transcriptome libraries were prepared for RNA sequencing. Because the RNA-Seq data are not yet available, I analyzed a related data set from a long-term selection experiment in which the survival and performance of C. virescens on a novel, highly defended, host plant (Physalis angulata) was examined. In comparing the genomes of selection line and control line C. virescens, I found variation in some Cytochrome p450 genes. This gene family has a demonstrated role in allowing some herbivorous insects to feed on highly defended hosts, and has been implicated in insecticide resistance. I found amino acid level variation between selection and control lines in the gene region that is responsible for substrate binding, suggesting that selection line caterpillars may have an improved ability to cope with the defense compounds of P. angulata. Future analysis of the transcriptome data from my recent bioassays will help elucidate whether Cytochrome P450s are intimately involved in the ability of C. virescens to feed upon chemically diverse hosts.
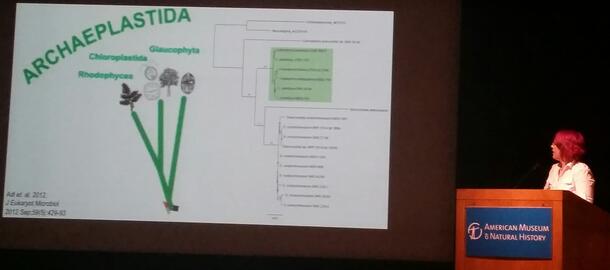
Transmission Electron Microscopy of Cyanophora paradoxa to Create 3D Reconstructions of Cytoskeletal Features
Kaleigh Lukacs (Pennsylvania State University)
Mentors: Aaron Heiss and Eunsoo Kim, Division of Invertebrate Zoology
Cyanophora paradoxa is a biflagellate alga belonging to the Glaucophyta subgroup of Archaeplastida. Since its discovery, no extensive imaging work has been done to map its major cytoskeletal structures, such as basal bodies (centriole homologues that anchor and give rise to the flagella) and roots (long connected series of microtubules, which associate with a specific basal body and span the cell to support it). These complexes are useful for evolutionary comparison, as their similarities and differences give supplementary support to molecular phylogenetic hypotheses. To efficiently document these structures, 3D reconstructions are created from transmission electron microscope (TEM) images of thin sections of the cells. Features in each section are manually annotated in an illustration program with representative geometry, then imported into a computer-aided drawing (CAD) program that assembles the sections. The resulting 3D model is a convenient, visually intuitive system for comparing its cytoskeleton to those of other organisms. In the case of Cyanophora, comparisons will be made amongst cells of other eukaryotic lineages; cytoskeletal data will be invaluable for its accurate placement into phylogenetic trees.
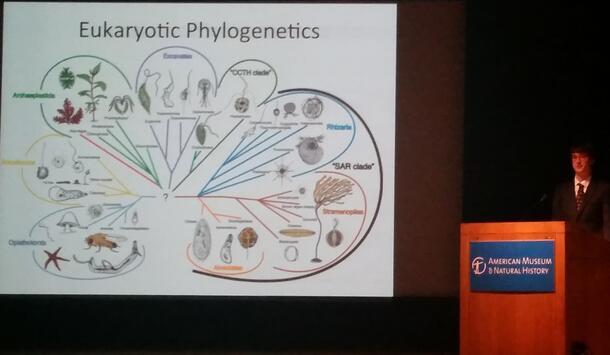
Compartmentalization: Testing a Tool for Phylogenetic Inference of Large, Messy Data Sets
Matthew Farnitano (Duke University)
Mentors: Aaron Heiss and Eunsoo Kim, Division of Invertebrate Zoology
Attempts to reconstruct the deep evolutionary history of eukaryotes have repeatedly produced inconclusive or conflicting results, partly due to the highly divergent and noisy nature of the available data. While a number of ‘supergroups’ have emerged with relatively strong support, the relationships between them are still contested. One method of resolving these relationships is large-scale phylogenomic analysis using many genes. This method increases the total phylogenetic signal of the data, but is often plagued by poorly aligned regions and missing data. Missing data may be the result of deletions, truncation, or the complete loss of certain genes in a lineage, as well as a lack of sequencing coverage in lesser-studied taxa. A new procedure termed compartmentalization has the potential to reduce the possible detrimental effects of missing data, while retaining phylogenetic information. In this procedure, subgroups known with confidence to be monophyletic are chosen from the taxa, and rooted subtrees are generated. In the full dataset, each of these subgroups is replaced by a reconstructed ancestral sequence representative of the subgroup as a whole. A maximum likelihood analysis is run on the collapsed data, generating a backbone tree onto which the subtrees can be regrafted. By collapsing large groups into representative sequences, the procedure can remove gaps without discarding informative data, resulting in better alignments and, ideally, better final tree topologies. We present an automated framework for the compartmentalization of single-gene and multi-gene datasets, and determine its usefulness for phylogenomic analysis.